
1. What Are Oncolytic Viruses?
Cancer therapy has always been a balance between destruction and preservation—how to eliminate malignant cells without inflicting unbearable damage on healthy tissues. Traditional approaches like chemotherapy and radiation tilt heavily toward destruction, harming both targets and bystanders. Targeted therapies offer refinement but often succumb to resistance. Into this landscape has emerged a new class of biologics that takes advantage of the tumor’s own weaknesses: oncolytic viruses. And as these therapies move from discovery toward clinical reality, the role of the oncolytic virus CDMO has become central, ensuring that promising science can be translated into scalable, GMP-grade treatments that actually reach patients.
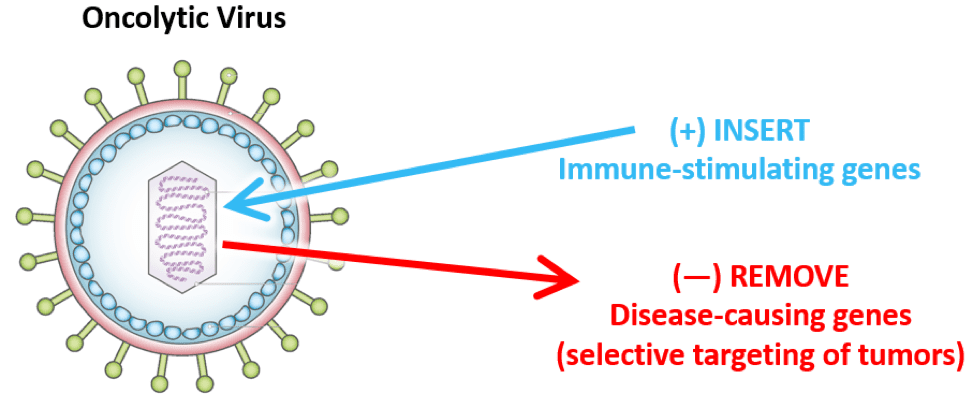
Oncolytic viruses (OVs) are either naturally occurring or genetically engineered viruses that selectively infect and replicate inside cancer cells. Unlike normal tissue, tumors often harbor defective antiviral defenses and dysregulated signaling pathways. These vulnerabilities make them permissive to viral replication, allowing OVs to amplify within tumors and cause direct lysis of cancer cells. Some viruses, like reovirus, already display this selectivity in nature. Others, such as adenovirus or herpes simplex virus, are engineered by deleting or altering genes that are essential for replication in healthy tissue but dispensable in cancer.
Modern genetic engineering allows researchers to go further, transforming OVs into multi-tasking cancer therapeutics. Their genomes can be armed with immune-stimulating payloads such as GM-CSF, checkpoint inhibitors, or bispecific engagers designed to draw immune cells into the tumor microenvironment. By doing so, OVs act not just as direct killers of cancer cells but as living delivery vehicles that reshape the immune response.
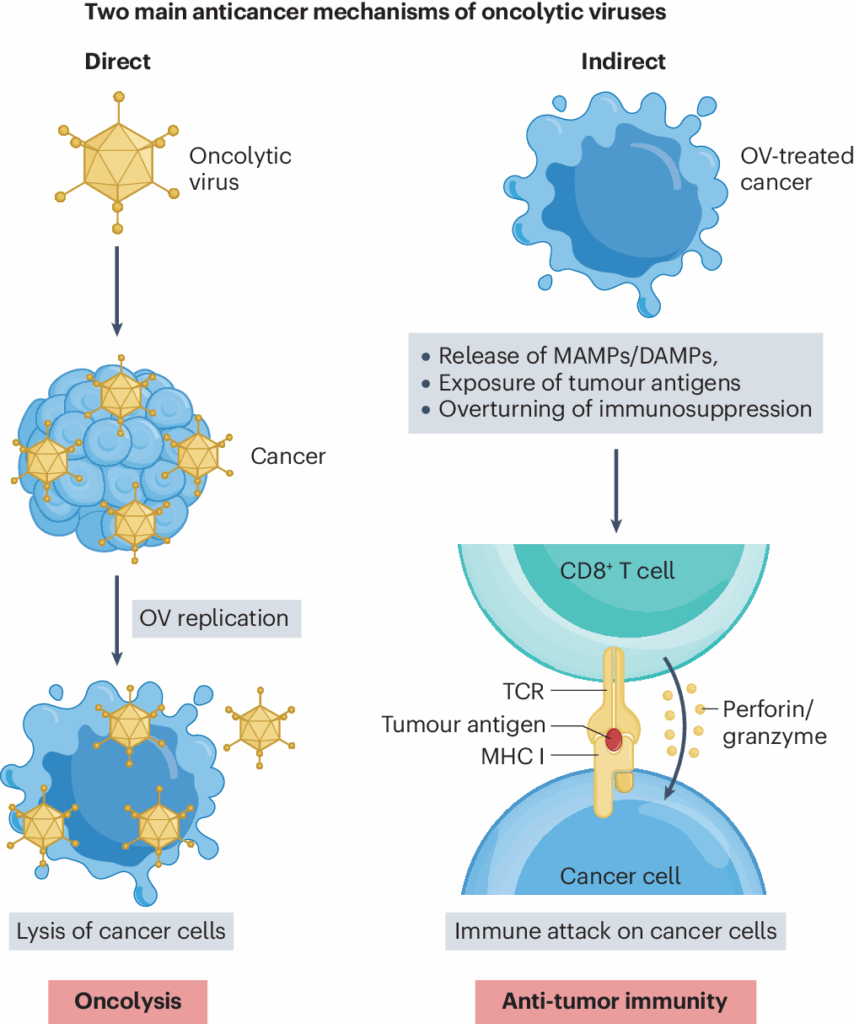
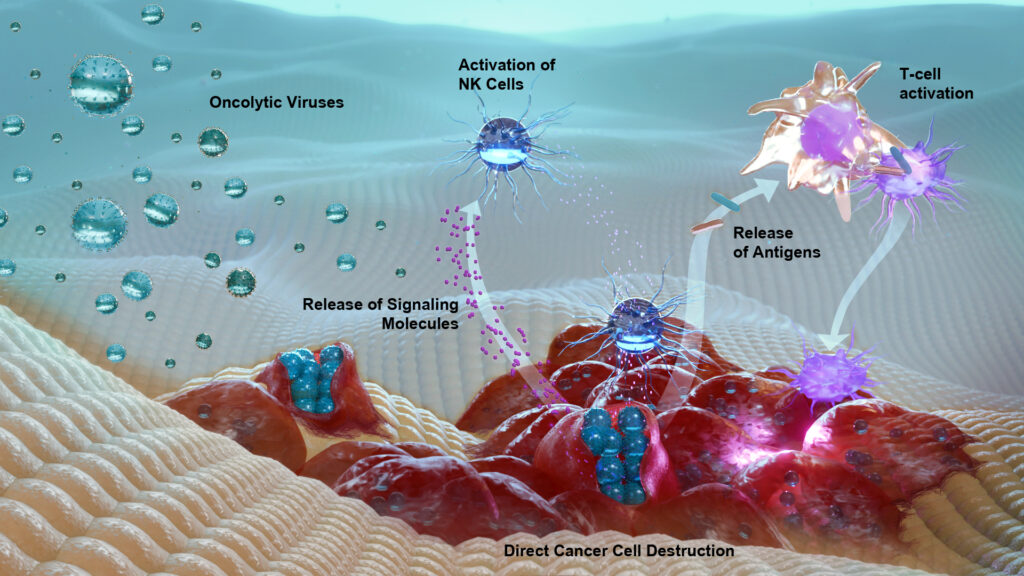
The power of oncolytic viruses lies in their synergistic effects. First, they cause direct tumor destruction through viral replication and cell lysis, releasing new infectious particles to propagate within the tumor mass. Second, this death is immunogenic, spilling tumor antigens and viral components that serve as a rallying cry for the immune system. Dendritic cells capture and present these antigens, priming T cells to mount a systemic, long-lasting anti-tumor response. In effect, OVs can turn a tumor into its own personalized cancer vaccine. For this potential to become a clinical reality, however, programs must move from scientific promise to GMP execution, and that is where choosing the right oncolytic virus CDMO makes the difference between a breakthrough therapy and an idea that never leaves the lab.

Their compatibility with other immunotherapies makes them even more valuable. Cold tumors, which resist checkpoint inhibitors due to poor immune infiltration, can be inflamed by OV infection, converting them into responsive lesions. The large genome capacity of certain viral backbones also allows them to carry complex payloads, combining oncolysis, immune stimulation, and targeted delivery in one therapeutic package.
Multiple viral families are under active development, each with its own strengths. Adenoviruses are attractive for their engineering flexibility and production scalability. Herpes simplex virus provides room for large payloads. Vaccinia virus replicates rapidly and avoids integration into host DNA by staying in the cytoplasm. Vesicular stomatitis virus offers potent lytic activity and innate immune stimulation. Reovirus continues to be a model for natural tumor selectivity. Each backbone presents trade-offs in immunogenicity, safety, and manufacturability, which is why the OV pipeline is diverse and rapidly expanding.
This combination of direct oncolysis, immune activation, and genetic flexibility makes OVs one of the most promising frontiers in oncology. Yet, as the science moves from preclinical promise to clinical reality, challenges arise. Manufacturing these living biologics at GMP scale is vastly more complex than producing traditional drugs. In the next sections, we will explore why scaling oncolytic virus production is uniquely challenging, how specialized oncolytic virus CDMOs have emerged to bridge this gap, and what capabilities define the best partners for bringing OV programs from bench to bedside.
2. Why Manufacturing Oncolytic Viruses Is So Challenging
Moving from discovery science to clinical-grade production is where many oncolytic virus programs stumble. Unlike small molecules, which can be chemically synthesized with precision, or even monoclonal antibodies, which are produced through relatively standardized bioprocessing platforms, viruses are inherently complex living entities. They replicate inside host cells, and their behavior is shaped by the biology of both the virus and the host system. This dual complexity creates significant challenges in manufacturing.
First, host system requirements vary. OVs must be grown in specific mammalian cells that support their replication, such as HEK293, Vero, A549, or BHK21 cells. Each virus has its own host preferences, and each cell line brings its own regulatory considerations. For example, Vero cells are widely used and accepted by regulators, but they require stringent adventitious agent testing. HEK293 cells are highly permissive but carry baggage around their origin. Matching the virus to the right host system and developing a scalable cell bank is a critical step that CDMOs must master.
Second, yields are notoriously variable. The replication kinetics of viruses depend on many factors: multiplicity of infection (MOI), cell density at infection, nutrient availability, and incubation times. Even slight deviations in culture conditions can cause dramatic swings in viral titers. This makes process characterization and control strategies essential. Without careful development, a process that works at 2 liters in a lab may fail catastrophically at 200 liters in a GMP bioreactor.
Purification is another major hurdle. Viruses are delicate particles. Methods like ultracentrifugation, while useful at research scale, shear viral particles and cannot scale. Chromatographic methods such as ion exchange or size exclusion can separate viruses from host proteins and DNA, but each must be tailored to the specific virus to avoid loss of infectivity. The balance between purity and potency is difficult—removing contaminants while preserving intact, infectious virus is a tightrope walk.
Safety cannot be overstated. Because viruses are replicating entities, they require strict biosafety and GMP containment. Facilities must maintain segregation between viral programs, validated viral inactivation procedures, and thorough cleaning validation to prevent cross-contamination. Every batch must be tested for sterility, mycoplasma, and adventitious viruses. Regulators scrutinize these programs heavily, and a single contamination event can destroy credibility.
Potency assays represent yet another challenge. Counting particles or measuring viral genomes is insufficient. Regulators demand functional assays that measure what the virus actually does—infect and kill tumor cells. These assays are complex, time-consuming, and highly variable. Developing a reproducible potency assay linked to the virus’s mechanism of action is one of the most difficult tasks in OV development.
Finally, scalability remains the overarching issue. Many labs can produce research-grade OVs at small volumes, but scaling to 500 or 1,000 liters while maintaining consistency, safety, and potency is extremely difficult. This is why specialized CDMOs with viral expertise exist—because without them, the vast majority of programs will never reach clinical supply. For innovators determined to advance into the clinic, working with a proven oncolytic virus CDMO is the surest way to overcome these barriers and translate groundbreaking science into a viable therapeutic reality.
3. The Role of Oncolytic Virus CDMOs
An oncolytic virus CDMO is not just a contract manufacturer—it is a strategic partner that enables translation from academic discovery to regulatory-approved clinical programs. What sets these CDMOs apart is their combination of scientific, engineering, and regulatory expertise, tailored specifically to the challenges of live viral therapeutics.
The first role of an oncolytic virus CDMO is process development. They design upstream production methods that balance yield with quality, and downstream purification processes that achieve high purity without sacrificing infectivity. These processes must be robust, reproducible, and compliant with GMP requirements. A strong CDMO develops them with scale-up in mind from day one.
The second role is viral safety. CDMOs build facilities with segregation to prevent cross-contamination, validated viral inactivation methods, and biosafety controls aligned with BSL-2 or BSL-3 requirements depending on the virus. They also manage comprehensive adventitious agent testing and environmental monitoring programs to satisfy regulators.
Analytics is another core contribution. Top CDMOs develop and validate assays for infectivity, potency, genome quantification, purity, and identity. They ensure these assays are not just research tools but are suitable for release testing under GMP.
Manufacturing under GMP conditions is, of course, the centerpiece. CDMOs produce Phase I, II, and III clinical lots with full traceability, batch records, and quality oversight. They maintain electronic systems for data integrity, manage master and working cell banks, and support comparability studies when processes change.
Beyond production, CDMOs provide regulatory guidance. They help author CMC sections of IND or IMPD filings, design comparability protocols, and prepare for regulatory inspections. Many biotechs lack this in-house expertise, making CDMOs indispensable for successful filings.
Fill–finish is another capability. Oncolytic viruses must be filled aseptically into vials or pre-filled syringes. This requires specialized viral fill–finish lines with low-shear pumps, isolators, and validated container–closure systems.
Finally, top CDMOs manage lifecycle needs: technology transfer between facilities, scale-up to commercial volumes, and long-term supply chain management. They become an extension of the client’s development team, providing continuity and expertise that persists from plasmid DNA all the way to clinical-grade virus.
In essence, the best oncolytic virus CDMOs serve as strategic gatekeepers. Without them, most OVs would remain trapped in academic labs or preclinical development. With them, OVs become viable clinical products capable of reshaping cancer therapy.
4. Process Development for Oncolytic Viruses
Manufacturing oncolytic viruses is fundamentally different from producing antibodies or small molecules. Viruses are living, replicating entities that require a suitable host system and conditions that allow replication while maintaining safety and consistency. This makes process development the most critical stage in any OV program. A well-designed process ensures that yields are reproducible, purity is acceptable to regulators, and potency is maintained from discovery through GMP scale-up.
4.1 Upstream: Viral Production
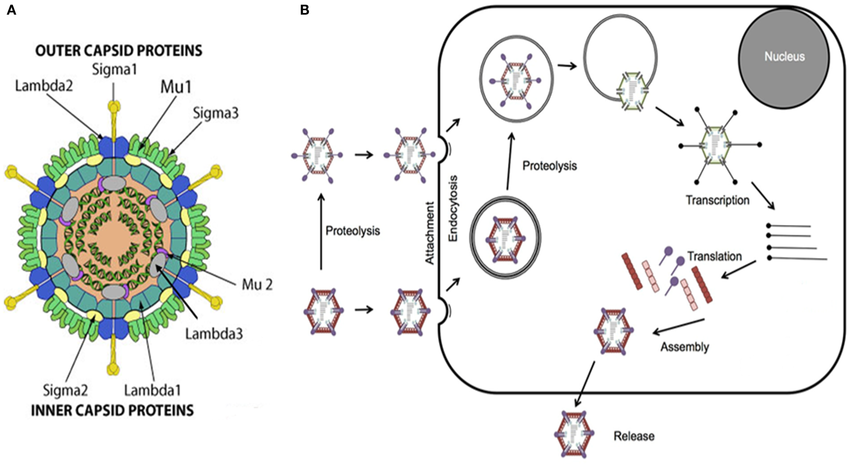
Cell line choice is the first major variable. Oncolytic viruses are grown in mammalian cells, and the optimal host depends on the virus backbone. Vero cells are widely used and regulator-accepted for herpes simplex virus (HSV) and vaccinia. HEK293 cells are preferred for adenovirus, where suspension-adapted HEK293 cultures allow for efficient, scalable production. Other lines like A549, BHK21, or proprietary GMP-qualified cell banks are also in play. The choice influences yields, regulatory acceptance, and even the eventual cost of goods.
Culture mode is another key factor. Suspension systems are the gold standard for scalability, as they can be adapted to single-use stirred tank bioreactors. Adherent systems are still required for viruses like HSV that have not been adapted to suspension. In these cases, microcarriers or fixed-bed systems expand the surface area available for cell attachment, enabling large-scale growth.
Infection parameters, such as multiplicity of infection (MOI), timing of infection, and harvest window, are carefully optimized for each virus. A low MOI can improve amplification efficiency but may lengthen production time. The harvest window must be chosen to maximize viral titers while avoiding excessive host cell lysis that can complicate purification.
The use of serum-free, chemically defined media is becoming standard. Serum-free systems reduce batch-to-batch variability, eliminate risks of adventitious agent contamination from animal-derived products, and simplify regulatory approval.
Scalability is the ultimate test of upstream process development. Early work may begin in T-flasks or roller bottles, but processes must be engineered to transition smoothly into 2–10 liter bench bioreactors, 50–200 liter pilot systems, and finally 500–2,000 liter GMP runs. For adenoviruses, HEK293 suspension cultures in chemically defined media have become the benchmark. HSV still frequently requires adherent Vero cells, which demands more specialized equipment. Each virus requires a tailored, bespoke solution.
4.2 Downstream: Purification
If upstream is about growth, downstream is about survival. Oncolytic viruses are far more fragile than antibodies or recombinant proteins. The downstream challenge is to remove contaminants while maintaining viral infectivity.
Clarification is the first step after harvest. Cell debris and large particles are removed through depth filtration or microfiltration. This step must be optimized to reduce fouling while maintaining virus recovery.
Concentration and diafiltration are typically carried out using tangential flow filtration (TFF). Membranes are carefully selected to retain viral particles while allowing smaller contaminants to pass through. Operating conditions—such as pressure, flow rate, and buffer composition—are finely tuned to minimize shear damage to the virus.

Chromatography plays a central role in purification. Ion exchange chromatography can separate viruses from host cell DNA and proteins based on charge differences. Size exclusion chromatography can further purify virus particles from aggregates. Affinity methods are also used when specific capture ligands exist. Each method must be validated for each virus to confirm that infectivity is preserved.
Ultracentrifugation, a staple of research labs, cannot scale. It is labor-intensive, prone to variability, and damaging to viruses under high g-forces. Scalable alternatives like chromatography and TFF are therefore the backbone of GMP processes.
Polishing steps focus on removal of host cell proteins, DNA, and other impurities to levels acceptable to regulators. Residual DNA must be reduced to picogram-per-dose levels, and host protein contamination must be minimized. The challenge is always to achieve these purity levels without compromising viral potency.
The overarching challenge is balance: achieving high recovery while maintaining intact, infectious viral particles. A top-tier CDMO brings not only the equipment but also the accumulated know-how to optimize this balance.
5. Analytics and Quality Control
Analytics are the language regulators speak, and for oncolytic viruses, particle counts alone will never satisfy them. A comprehensive quality control (QC) strategy is required to demonstrate identity, purity, potency, and safety, and this is precisely where the expertise of an experienced oncolytic virus CDMO becomes critical.
Identity testing confirms that the virus is the intended product. This involves genome sequencing, PCR assays to verify engineered modifications, and protein analysis of viral capsid or envelope components.
Purity testing focuses on removing what should not be in the product. Host cell proteins, residual host DNA, and serum-derived proteins must be quantified and reduced below regulatory thresholds. Process-related contaminants from media, reagents, or purification steps must also be eliminated.
Potency assays are among the most complex. Regulators require functional, cell-based assays that reflect the virus’s actual mechanism of action. These may include plaque assays to measure replication, TCID50 assays for infectious dose, or cytopathic effect assays to track cancer cell death. Since these assays are variable by nature, reproducible and validated methods are essential.
Titer determination combines genome copy counts (via qPCR or ddPCR) with infectious unit measurements (PFU/ml). Both are required, since genome counts alone cannot distinguish infectious from non-infectious particles.
Safety assays are non-negotiable. Every batch undergoes mycoplasma testing, sterility checks, adventitious virus screens, and endotoxin assessments.
Finally, stability testing defines how long a product remains viable under real-world conditions. Oncolytic viruses must be evaluated under freeze–thaw cycles, various storage temperatures, and ICH stability protocols to establish shelf life and handling requirements.
A strong oncolytic virus CDMO integrates all of these assays into validated QC workflows. This ensures data are acceptable to regulators, safeguards patient safety, and locks in a process robust enough to support clinical development and eventual commercial supply.
6. GMP Considerations for Oncolytic Viruses
6.1 Biosafety and Containment
Manufacturers must handle oncolytic viruses at the correct biosafety level. Depending on the viral backbone and any modifications, this work may require BSL-2 or BSL-3 facilities. Teams maintain strict segregation between programs to eliminate the risk of cross-contamination. Operators complete specialized training in viral vector handling, gowning, and aseptic technique before they can enter production areas. Environmental monitoring runs continuously, and teams validate containment systems on a routine schedule.
6.2 GMP Compliance
GMP compliance extends far beyond cleanroom classification. A strong quality management system aligns with ICH Q5 and Q6 guidance for biologics. Production staff document every step of the process in batch records that allow full traceability. Electronic data systems protect integrity and prevent tampering. Teams validate cleaning and sterilization methods and prove viral inactivation procedures for waste handling. GMP compliance requires a culture of discipline and accountability at every level.
6.3 Fill–Finish
Fill–finish is often the step that determines whether a product succeeds or fails. Oncolytic viruses are highly shear-sensitive, so teams use aseptic filling under Grade A isolators with low-shear pumps to protect product integrity. Final presentations include both vials and pre-filled syringes, depending on clinical needs. Teams perform container–closure integrity testing, analyze extractables and leachables, and conduct stability studies under real-world conditions to verify that the final drug product remains safe and effective.
7. Regulatory Landscape
Regulatory agencies expect sponsors to provide comprehensive CMC data packages for oncolytic viruses. Developers must demonstrate identity, purity, potency, and safety through validated assays. When scaling or modifying a process, teams must perform comparability studies to prove the product remains equivalent. Regulators also insist that potency assays link directly to the mechanism of action rather than relying on surrogate markers. Developers must provide risk assessments for immunogenicity, biodistribution, and viral shedding, particularly for novel virus platforms.
Both the FDA and EMA classify oncolytic viruses as biologics, but each agency approaches the details differently. CDMOs with experience in the space anticipate these questions and align processes with global standards. The most effective partners also support clients directly in pre-IND or Scientific Advice meetings, ensuring regulators view the program with confidence.
8. How Oncolytic Virus CDMOs Differentiate Themselves
Not all CDMOs are equal. The best distinguish themselves through specialization and infrastructure. Viral vector specialization matters—general biologics experience does not guarantee viral expertise. Facilities must have BSL-2/3 environments, single-use bioreactors, and dedicated viral fill–finish lines. Experience with multiple viral backbones such as adenovirus, HSV, vaccinia, reovirus, and VSV builds credibility.
Analytical innovation is another differentiator. CDMOs that can provide potency assays acceptable to regulators give clients an advantage. Regulatory integration—support with CMC authoring, IND/IMPD submissions, and regulatory meetings—is critical. Finally, speed matters. The best CDMOs can move a program from R&D to IND in approximately 12 months when platforms are in place.
9. Why Partner Early With an Oncolytic Virus CDMO
Engaging a CDMO early ensures that processes are designed for manufacturability from the beginning. Potency, identity, and safety assays can be developed and validated early, rather than retrofitted later. Regulatory alignment is achieved sooner, and comparability strategies are built into the development plan. Companies that delay CDMO engagement often face rework, delays, and regulatory setbacks that could have been avoided.
10. The Future of Oncolytic Virus CDMOs
The field of oncolytic viruses is expanding rapidly. As immunotherapy combinations become more common, OVs are poised to play multiple roles: priming the immune system, directly lysing tumors, and delivering therapeutic genes. With dozens of candidates in Phase I/II trials and several approaching Phase III, the demand for experienced CDMOs will surge.
The CDMOs that succeed will be those that specialize in viral vectors, offer end-to-end solutions from plasmid through fill–finish, integrate regulatory foresight into every step, and build scalable processes that withstand global scrutiny.
Conclusion: Oncolytic Virus CDMOs as Strategic Gatekeepers
Oncolytic viruses represent a rare convergence in oncology: a therapy that kills tumors from within while transforming them into immune beacons for the body to attack. They sit at the intersection of virology, immunology, and genetic engineering—a frontier science with extraordinary promise. Yet promise alone does not save patients. A brilliant virus that cannot be manufactured, scaled, or validated under GMP is simply another lab experiment.
This is where oncolytic virus CDMOs redefine the playing field. They are not background vendors; they are the architects of translation, the ones who turn fragile research systems into reproducible processes, who weave analytics into regulatory narratives, and who build GMP supply chains capable of carrying these therapies into Phase I, II, and beyond. Without them, the field stalls. With them, oncolytic virotherapy becomes a legitimate pillar of cancer treatment.
For biotechs pushing forward in this space, the decision is not about outsourcing—it is about partnership. Selecting the right oncolytic virus CDMO is less a procurement exercise and more a strategic alliance, one that will decide whether a program advances to patients or dies on the bench. The science may ignite the spark, but the CDMO partnership carries the flame to the clinic
13 Oncolytic Virus CDMO FAQs
1. What exactly is an oncolytic virus, and why should I care?
Imagine a therapy that sneaks into tumors, replicates like mad, and blows cancer cells apart from the inside—then calls in the immune system to finish the job. That’s the allure of OVs: they’re both assassin and whistleblower.
2. Why can’t my lab just scale up OVs on its own?
Because growing a live virus for patients isn’t like brewing craft beer. The stakes are higher, the regulators are stricter, and one slip can turn your beautiful virus into a regulatory horror story. CDMOs exist to keep the dream alive and compliant.
3. What do oncolytic virus CDMOs actually do?
Think of them as the guardians of translation. They design, test, and manufacture your virus so it doesn’t collapse under GMP scrutiny. Upstream? Handled. Downstream? Polished. Analytics? Validated. Fill–finish? Aseptic and flawless.
4. Which cell lines are most common?
HEK293 for adenovirus. Vero for HSV. Each backbone has its muse. The choice isn’t just biology—it’s regulatory pedigree, scalability, and yield optimization.
5. Why is purification such a pain?
Because viruses are divas. Push them too hard through filters and they fall apart; treat them too gently and impurities cling like groupies. Mastering purification is part science, part art.
6. How do regulators look at potency assays?
Particle counts won’t cut it. Regulators want to see what your virus does: infect, replicate, kill tumor cells, and spark immune activation. A good CDMO builds assays that prove function, not just form.
7. What makes GMP production so intense?
Segregated suites, biosafety validations, and operators trained like elite athletes. Every door you open, every filter you swap, every record you sign is part of a choreography regulators expect to see—and inspect.
8. Can oncolytic viruses be combined with other therapies?
Absolutely. They play especially well with checkpoint inhibitors, CAR-Ts, and radiation. But to get there, you need comparability studies, validated formulations, and a CDMO that can support combo trial demands.
9. What’s the timeline from R&D to IND?
With a ready platform and the right CDMO, about 12 months. Without? Let’s just say “years” and “burn rate” in the same sentence.
10. Why is fill–finish so overlooked?
Because people underestimate how fragile viruses are. A low-shear fill process under Grade A isolators is the difference between stable vials and dead product. Sexy? Maybe not. Critical? Always.
11. What role do analytics really play?
Analytics are your passport. Genome copies, infectious units, purity panels, stability data—without them, your IND gets stamped “denied.” A sharp CDMO knows how to make regulators nod, not frown.
12. What mistakes kill OV programs fastest?
Using academic ultracentrifugation past discovery. Treating potency as an afterthought. Delaying CDMO engagement until the cliff’s edge. Each mistake costs time, money, and credibility.
13. Why does choosing the right CDMO feel like dating?
Because it is. You’re not just buying capacity—you’re trusting a partner with your most vulnerable asset. The right oncolytic virus CDMO understands your science, anticipates your risks, and stays with you through the messy, thrilling, expensive, exhilarating journey from bench to bedside. Pick wrong, and heartbreak (and FDA hold letters) follow.
Want to learn more? Click here–> Oncolytic Virus CDMO Services
Want to contact our sales team? Email: info@elisebiopharma.com