
Biologics are changing.
For years, the industry has focused on proteins, monoclonal antibodies, and other relatively stable molecular systems. Those products are still complex, but the manufacturing logic is familiar: express the molecule, purify it, characterise it, and stabilise it.
That logic is starting to break.
Phages, live biotherapeutics, and exosomes are moving into clinical and commercial focus, and they do not behave like static molecules. Phages replicate through host interaction. Live biotherapeutics remain metabolically active. Exosomes exist as heterogeneous vesicle populations shaped by the cells that produce them. In each case, the therapy cannot really be separated from the biological system behind it.
That creates a very different manufacturing challenge.
Success is no longer just about making the same molecule again and again. It is about controlling biological systems that are variable, responsive, and sensitive to process conditions. Small changes in fermentation, cell culture, purification, or formulation can affect not only yield, but function. That is exactly why more sponsors are looking for a phage live biotherapeutic exosome CDMO rather than a conventional manufacturing partner.
The pattern is becoming familiar across advanced biologics. Early data looks strong. Proof of concept works. Then scale-up begins, variability increases, stability becomes harder to maintain, and reproducibility starts to slip. Programmes do not stall because the science stops being interesting. They stall because the process stops being controlled.
That is why demand is growing for a phage live biotherapeutic exosome CDMO with real depth in upstream development, downstream processing, stabilisation, and analytical strategy. Across phage manufacturing, live biotherapeutic manufacturing, and exosome production, the same rule applies: the process defines the product.
Upstream, downstream, and formulation cannot be treated as separate boxes. Stabilisation is not a final add-on. Analytical methods have to measure function, not just structure. Manufacturing becomes an integrated system rather than a simple chain of operations.
This is where differentiation starts to matter.
The best CDMO is not defined by capacity alone. It is defined by the ability to control complex biological systems across their full lifecycle. Elise Biopharma is built around that requirement. Rather than treating phages, live biotherapeutics, and exosomes as isolated service areas, the company brings microbial fermentation, particle and vesicle systems, and formulation and stabilisation together in one development framework.
As these modalities advance, manufacturing is no longer a downstream consideration. It is increasingly the limiting factor. And the phage live biotherapeutic exosome CDMO that can control complexity, not just produce volume, will be the one that helps programmes succeed.
The Convergence of Three Complex Modalities
The current generation of biologics is not defined by incremental improvements in protein engineering or antibody optimization. It is defined by a structural shift in what constitutes the “drug” itself. Phage therapeutics, live biotherapeutic products (LBPs), and exosomes are not simply new categories—they represent a fundamental change in how therapeutic systems behave, how they are manufactured, and how they fail.
At a surface level, these modalities appear unrelated. Phages are viral entities that infect bacteria. Live biotherapeutics are living microbial systems designed to interact with host physiology. Exosomes are nanoscale vesicles that mediate intercellular communication. Each operates in a different biological domain, with different regulatory considerations and different clinical endpoints.
Yet from a manufacturing perspective, they converge with surprising precision.
Living Systems Versus Inert Biologics
Traditional biologics—monoclonal antibodies, recombinant proteins, and even many vaccines—are ultimately static outputs. A protein is expressed, purified, and stabilized. Its structure is fixed. Its function is largely determined by sequence and folding. Manufacturing is complex, but the end product is chemically and structurally defined.
Phages, LBPs, and exosomes do not behave this way.
- Phages replicate. Their yield depends on infection kinetics, host cell physiology, and environmental conditions.
- Live biotherapeutics remain metabolically active. Their viability, gene expression, and interaction with the host continue beyond manufacturing.
- Exosomes are secreted particles. Their composition depends on the upstream cellular environment and cannot be reduced to a single molecular identity.
In all three cases, the “product” is not fully separable from the biological system that produces it.
This introduces a critical shift:
manufacturing is no longer about producing a defined molecule—it is about controlling a biological system within acceptable bounds.
Process Defines Product
In conventional biologics, process optimization improves yield, purity, and cost efficiency. In emerging biologics, process changes can redefine the product itself.
This is most evident in exosome manufacturing. Small changes in:
- culture conditions
- media composition
- stress signals
- cell density
can alter vesicle size distribution, surface markers, and cargo composition. Two batches produced under slightly different conditions may not be functionally equivalent, even if they meet basic characterization criteria.
The same principle applies to phages. Amplification conditions influence:
- burst size
- phage morphology
- host contamination levels
- endotoxin burden
A change in host strain or growth phase can produce materially different outputs.
For live biotherapeutics, fermentation parameters determine:
- strain viability
- metabolic state
- expression of engineered pathways
- stability during downstream processing
This leads to a governing reality:
In phages, LBPs, and exosomes, the process is not a pathway to the product. The process is the product.
A CDMO operating in this space must therefore treat upstream, downstream, and formulation as a single continuous system, not as modular steps.
Variability Is Not Noise—It Is the Core Problem
All biologics manufacturing deals with variability. But in traditional systems, variability is often statistical—batch-to-batch differences that can be minimized and controlled.
In emerging modalities, variability is biological and structural.
- Exosomes are inherently heterogeneous populations of vesicles.
- Phage preparations can contain mixtures of active and inactive particles.
- Live biotherapeutics can shift in viability and phenotype depending on environmental stress.
This is not a deviation from an ideal state—it is intrinsic to the system.
As a result, quality control cannot rely solely on conventional metrics such as purity or concentration. It must incorporate:
- functional assays
- potency measurements
- viability thresholds
- particle characterization
The analytical burden increases significantly, and the definition of “release-ready” material becomes more complex.
Why Traditional CDMOs Struggle
Most established CDMOs are optimized for:
- monoclonal antibody production
- recombinant protein expression
- standard viral vector manufacturing
These platforms assume:
- a stable molecular product
- well-defined purification pathways
- established regulatory frameworks
When applied to phages, LBPs, or exosomes, these assumptions begin to break down.
Common failure points include:
1. Over-reliance on platform thinking
Standardized processes are applied to systems that require customization at every stage.
2. Insufficient upstream control
Fermentation or cell culture is treated as a yield problem rather than a product-definition problem.
3. Fragmented development pipelines
Upstream, downstream, and formulation are handled as separate domains, leading to mismatches in scale-up and stability.
4. Limited analytical depth
Conventional assays are insufficient to capture functional variability.
The result is predictable:
programs stall during scale-up, exhibit inconsistent performance, or fail to translate from research to GMP production.
The Need for Integrated Biological Control
The convergence of phages, live biotherapeutics, and exosomes reveals a single underlying requirement:
control across dynamic biological systems.
This control operates across three layers:
- Upstream system design
- host selection
- strain engineering
- culture conditions
- Downstream processing
- purification strategies
- removal of contaminants
- preservation of functional integrity
- Formulation and stabilization
- lyophilization
- encapsulation
- storage conditions
Failure at any one layer propagates through the system. Success requires that all three are developed in coordination.
This is where the definition of a “best CDMO” changes.
It is no longer sufficient to provide capacity or even technical capability in isolated areas. The leading CDMO in this domain must operate as a systems integrator, capable of aligning biological behavior, process engineering, and formulation science into a single, controlled framework.
Convergence as a Commercial Signal
The increasing volume of programs in phages, LBPs, and exosomes is not coincidental. It reflects a broader shift in therapeutic strategy:
- targeting microbiome interactions
- addressing antimicrobial resistance
- enabling intracellular delivery
- leveraging natural biological communication systems
These approaches require modalities that are inherently complex and difficult to manufacture.
The convergence of these modalities, therefore, is not just a scientific observation.
It is a commercial signal:
The future of biologics manufacturing will be defined by the ability to control complex, dynamic, and heterogeneous biological systems at scale.
Phages, live biotherapeutics, and exosomes are the first large wave of this transition. They are not edge cases—they are leading indicators.
And they impose a new standard on CDMOs:
not just the ability to produce, but the ability to control.
Phage Therapeutics: Why Phage Manufacturing Requires a Specialist CDMO
Phage therapeutics are reshaping advanced biologics, but phage manufacturing is far more complex than standard protein production. Unlike recombinant proteins or monoclonal antibodies, phages are amplified through infection, so yield, potency, and consistency depend on host cell health, replication kinetics, and tightly controlled process conditions.
That makes phage manufacturing a system-level challenge. A phage CDMO must manage host-derived impurities, endotoxins, cellular debris, and residual nucleic acids without damaging phage integrity. Endotoxin removal is especially critical in GMP phage manufacturing because bacterial amplification can generate impurity burdens far above those seen in other biologics platforms.
Purification is equally demanding. Filtration, chromatography, and centrifugation must be tailored to each phage’s size, morphology, and stability profile. There is no universal platform. Every phage therapeutics program requires customised upstream development, downstream purification, and formulation planning to preserve infectivity and improve recovery.
As phage therapeutics expand in antimicrobial resistance, oncology, and animal health, the need for an experienced phage CDMO is becoming more urgent. The best CDMO for phage therapeutics is not simply the one with production capacity. It is the one that can control phage amplification, purification, GMP scale-up, and fill-finish as one integrated manufacturing process.
Live Biotherapeutics and Engineered Probiotics: Manufacturing Living Systems
If phage therapeutics demonstrate how biologics can behave as dynamic replication systems, live biotherapeutics and engineered probiotics extend that complexity into a different dimension: persistence. These are not transient agents. They are living organisms introduced into biological environments where they must survive, function, and in many cases adapt.
This distinction changes the manufacturing problem in a fundamental way. The objective is no longer to produce a biologically active entity and stabilize it long enough for delivery. The objective is to produce a system that remains viable, functional, and predictable across time, environments, and patient variability.
At first glance, live biotherapeutics appear deceptively simple. They are often described using familiar language—bacteria, strains, fermentation, colony-forming units. This framing can obscure the underlying complexity. Unlike traditional fermentation products, where the output is a metabolite or protein, here the organism itself is the product. Its physiological state at the moment of manufacture carries forward into storage, transport, and ultimately into the patient.
This introduces a central constraint:
viability is not a specification—it is a moving target.
A strain that performs optimally during fermentation may not survive downstream processing. A formulation that preserves viability during storage may alter metabolic activity. A delivery system that protects against gastric conditions may delay or suppress functional expression in the target site. Each stage imposes trade-offs, and those trade-offs must be resolved within a tightly integrated development strategy.
The complexity increases further when engineered systems are involved. Many modern live biotherapeutics incorporate genetic modifications designed to enhance functionality—CRISPR-edited pathways, inducible gene circuits, metabolite production modules, or targeted interaction mechanisms. These modifications introduce additional layers of instability.
Genetic constructs must remain intact across:
- multiple fermentation generations
- scale-up transitions
- storage conditions
- in vivo environments
Plasmid loss, mutation, or silencing can transform a functional therapeutic into an inactive or inconsistent product. Ensuring stability is not simply a matter of sequence verification. It requires process design that minimizes selective pressure, maintains appropriate growth conditions, and incorporates analytical methods capable of detecting subtle shifts in population structure.
The challenge becomes even more pronounced with anaerobic organisms. Many of the most promising live biotherapeutic candidates—particularly those targeting the gut microbiome—are strict anaerobes. These organisms are highly sensitive to oxygen exposure, which complicates every stage of manufacturing.
Upstream processing must be conducted in controlled anaerobic environments. Even minor oxygen intrusion can reduce viability or alter metabolic state. Downstream processing introduces additional risk, as harvesting, concentration, and drying steps often expose cells to conditions outside their optimal range. Maintaining an anaerobic chain of custody from fermentation through final formulation is technically demanding and operationally intensive.
Scaling these systems adds another layer of difficulty. Fermentation processes that perform reliably at small scale do not necessarily translate to larger volumes. Oxygen gradients, mixing dynamics, nutrient distribution, and waste accumulation all change with scale. For aerobic systems, these variables are challenging but manageable. For anaerobic or microaerophilic organisms, they can fundamentally alter growth behavior.
As a result, scale-up is not a simple matter of increasing vessel size. It requires re-optimization of process parameters, often involving iterative cycles of development. Failure to account for these changes can lead to reduced yields, altered strain performance, or complete process breakdown.
Downstream processing introduces its own set of constraints. Unlike protein purification, where the goal is to isolate a stable molecule, live biotherapeutic processing must preserve cellular integrity. Centrifugation, filtration, and washing steps must be calibrated to avoid mechanical stress or osmotic shock. Even minor deviations can result in significant viability loss.
Drying technologies, particularly lyophilization, play a central role in stabilizing these products. However, freeze-drying is not a neutral process. It imposes thermal and osmotic stresses that can damage cell membranes, disrupt metabolic pathways, and reduce long-term stability. The selection of cryoprotectants, optimization of freeze-drying cycles, and control of residual moisture are all critical variables.
Encapsulation adds another dimension. Many live biotherapeutics require protection from gastric acid and bile salts to reach their site of action. Enteric coatings, microencapsulation, and advanced delivery systems are used to address this challenge. These technologies must balance protection with release kinetics, ensuring that organisms are delivered intact and activated at the appropriate location.
Multi-strain formulations further complicate the landscape. Different organisms may have different growth requirements, stability profiles, and sensitivities. Co-formulating them introduces the risk of competitive interactions, differential viability loss, and unpredictable behavior over time. Designing stable, multi-strain products requires careful consideration of strain compatibility, formulation conditions, and storage environments.
Regulatory considerations add yet another layer. Live biotherapeutics occupy a complex position between pharmaceuticals and biologically derived products. Definitions vary across jurisdictions, and requirements continue to evolve. Demonstrating consistency, safety, and efficacy requires comprehensive characterization of both the organism and the manufacturing process. This includes not only identity and purity, but also functional performance and stability over time.
These challenges collectively redefine what is required from a CDMO. It is not sufficient to offer fermentation capacity or standard downstream processing. A capable partner must understand the organism as a system, anticipate how it will respond to each stage of the process, and design manufacturing strategies that preserve its intended function.
This requires integration across disciplines:
- microbial physiology
- genetic engineering
- process engineering
- formulation science
Each decision must be made in the context of the entire system. Upstream conditions influence downstream stability. Formulation choices feed back into strain selection. Analytical methods must capture not only static properties, but dynamic behavior.
The convergence of these factors explains why live biotherapeutics and engineered probiotics are emerging as one of the most demanding categories in biologics manufacturing. They sit at the intersection of biology and engineering, where small changes can have disproportionate effects.
At the same time, they represent one of the most promising frontiers in therapeutics. The ability to modulate the microbiome, deliver functional biological activity, and interact with host systems in a controlled way opens pathways that are inaccessible to conventional drugs.
Realizing that potential depends on manufacturing. Not as a downstream consideration, but as a central component of product design.
In this context, the role of the CDMO becomes strategic. It is not simply a service provider, but a co-developer of the system. The ability to control viability, maintain genetic stability, manage anaerobic processes, and design effective formulations determines whether a program advances or stalls.
Live biotherapeutics make one point clear:
when the product is alive, manufacturing is not about production—it is about preservation, control, and translation across environments.
Exosomes and Extracellular Vesicles: Manufacturing Heterogeneous Particle Systems
If phages are defined by replication dynamics and live biotherapeutics by viability, exosomes introduce a third form of complexity: heterogeneity at the particle level. They are not living organisms, but they are not static molecules either. They exist as populations of vesicles—distributed across size, composition, and function—whose identity is inseparable from the cellular systems that produce them.
This makes exosome manufacturing one of the most conceptually difficult areas in modern biologics. The challenge is not simply producing vesicles. It is defining what those vesicles are, ensuring that definition is consistent, and maintaining that consistency through scale.
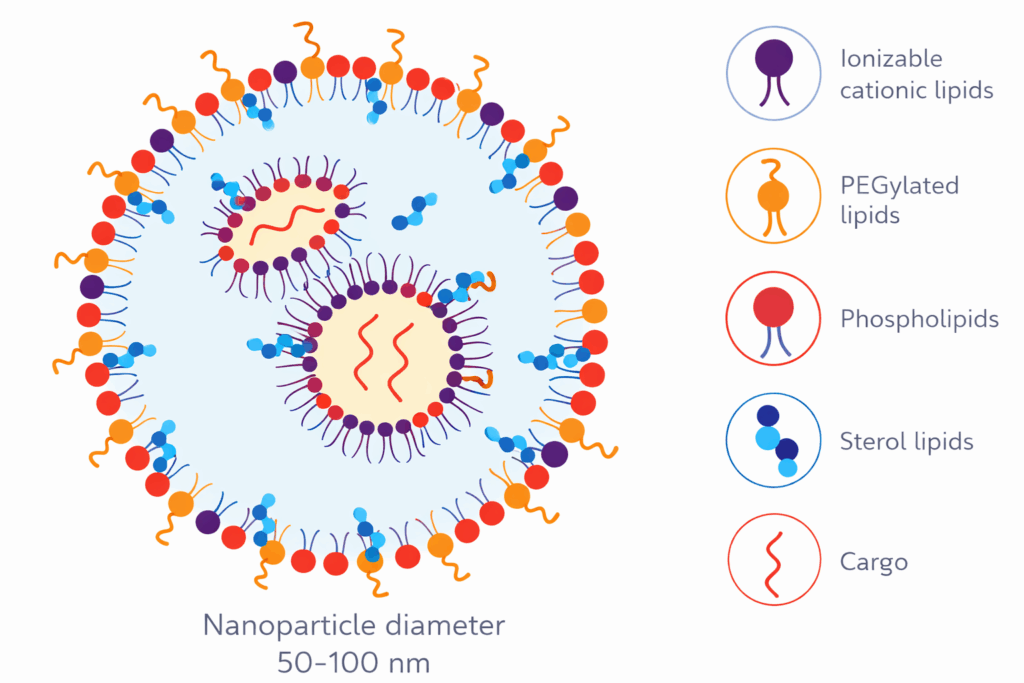
At a technical level, exosomes are extracellular vesicles typically ranging from 30 to 150 nanometers in diameter. They carry proteins, lipids, RNA, and other biomolecules derived from their parent cells. Their therapeutic potential stems from this ability to act as natural delivery systems—capable of transporting functional cargo across biological barriers, including, in some cases, the blood-brain barrier.
However, this same feature introduces a core constraint:
there is no single, fixed “exosome product.” There is only a distribution.
Each batch consists of a heterogeneous mixture of vesicles:
- varying in size
- differing in surface markers
- carrying different cargo compositions
- exhibiting different functional properties
Unlike a monoclonal antibody, which can be defined down to a precise molecular structure, exosomes must be defined statistically. Their identity emerges from population-level characteristics rather than a single entity.
This has immediate implications for manufacturing. The upstream process—cell line selection, media composition, culture conditions, and stress environment—directly shapes the vesicle population. Even subtle changes in these variables can shift the distribution of exosome size, composition, and function.
For example, altering nutrient availability or inducing mild cellular stress can change:
- vesicle secretion rates
- cargo loading patterns
- membrane composition
Two processes that appear similar on paper can produce vesicle populations with meaningfully different biological effects. This reinforces the principle introduced earlier: process defines product, but in exosomes, this relationship is particularly acute.
Upstream system design therefore becomes the first point of control. Selecting the appropriate cell source—whether stem cells, immune cells, or engineered cell lines—is not only a biological decision but a manufacturing one. Each cell type produces vesicles with distinct profiles, and those profiles must align with the intended therapeutic function.
Once vesicles are produced, the challenge shifts to isolation and purification. This is one of the most technically constrained areas in exosome manufacturing. Standard techniques such as ultracentrifugation, filtration, and chromatography each offer trade-offs between purity, yield, scalability, and structural integrity.
Ultracentrifugation, often used at research scale, can achieve relatively high purity but is difficult to scale and may damage vesicles through shear forces. Filtration-based approaches are more scalable but may allow co-isolation of unwanted particles. Chromatographic methods offer greater control but require careful optimization to avoid loss of functional vesicles.
No single method is sufficient across all programs. Instead, purification pipelines must be designed as integrated systems, combining multiple techniques to achieve acceptable performance. This again highlights the absence of a universal platform.
Characterization introduces another layer of complexity. Because exosomes are heterogeneous, analytical methods must capture distributions rather than single values. Techniques such as nanoparticle tracking analysis (NTA), dynamic light scattering (DLS), flow cytometry, and advanced imaging are used to assess size, concentration, and surface markers.
However, these methods provide only partial insight. Functional assays—measuring delivery efficiency, biological activity, or therapeutic effect—are often required to establish potency. This creates a gap between measurable physical properties and clinically relevant outcomes, which must be bridged through careful assay development.
Cargo loading adds further complexity, particularly for therapeutic applications involving RNA, proteins, or small molecules. Loading can occur endogenously, by engineering the parent cell, or exogenously, through techniques such as electroporation or chemical transfection. Each approach affects vesicle integrity, loading efficiency, and functional performance.
Stability is a persistent constraint throughout. Exosomes are sensitive to:
- temperature fluctuations
- freeze-thaw cycles
- mechanical stress
Aggregation, fusion, or degradation can alter their properties over time. Formulation strategies must therefore be developed to preserve vesicle integrity during storage and transport. This often involves buffer optimization, cryopreservation techniques, and, in some cases, exploration of lyophilization approaches—though freeze-drying exosomes introduces its own set of challenges.
Scaling exosome production amplifies all of these issues. Processes that are manageable at laboratory scale become significantly more complex when translated to bioreactors and GMP environments. Maintaining consistent vesicle populations across larger volumes requires tight control of upstream conditions and careful monitoring of process parameters.
This is compounded by the fact that exosome yield is often relatively low compared to other biologics. Achieving commercially viable production levels requires optimization not only of cell culture conditions but also of recovery efficiency. Losses at any stage—production, isolation, purification—can have a disproportionate impact on overall yield.
Regulatory frameworks for exosomes are still evolving, adding uncertainty to development pathways. Defining critical quality attributes, establishing potency assays, and demonstrating consistency across batches are active areas of discussion. This places additional pressure on manufacturing strategies to generate robust and defensible data.
From a CDMO perspective, these challenges converge into a single requirement: the ability to manage heterogeneity without losing control.
This requires:
- upstream expertise in cell systems and vesicle biology
- downstream capability in complex purification pipelines
- analytical depth to characterize distributions and function
- formulation knowledge to preserve integrity
Isolated capabilities are insufficient. An organization that excels in cell culture but lacks purification expertise will struggle to deliver consistent material. A group with strong analytical tools but limited upstream control will face variability at the source.
Exosome manufacturing therefore demands a systems-level approach similar to phages and live biotherapeutics, but with a different emphasis. Where phages require control of infection dynamics and LBPs require preservation of viability, exosomes require control of population structure.
This distinction is subtle but critical. The goal is not to produce identical particles, which is not feasible, but to produce populations that behave consistently within defined functional parameters.
As interest in exosome-based therapeutics continues to grow—driven by their potential in targeted delivery, regenerative medicine, and oncology—the pressure on manufacturing systems will increase. Programs that succeed will be those that can translate biological complexity into controlled, reproducible processes.
This reinforces the broader pattern emerging across all three modalities. Whether dealing with replicating viruses, living microbial systems, or heterogeneous vesicle populations, the central challenge is the same:
control over systems that resist simplification.
Exosomes represent the most abstract expression of this challenge. They are not defined by a single structure, but by a distribution shaped by biology. Manufacturing them is therefore not about producing a fixed entity. It is about shaping and maintaining a controlled biological outcome at scale.
The Hidden Bottleneck: Stabilization, Lyophilization, and Fill-Finish
Across phages, live biotherapeutics, and exosomes, the most consistent point of failure is not upstream biology or even purification. It is what happens after the product is made. Stabilization, formulation, and fill-finish are where otherwise viable programs lose functionality, reproducibility, or commercial feasibility.
This is not always obvious early in development. At laboratory scale, freshly prepared material often performs well enough to support proof-of-concept work. The system appears stable because it has not yet been exposed to real-world constraints—storage, transport, time, and environmental variability.
As programs move toward clinical or commercial stages, these constraints become unavoidable, and weaknesses in formulation strategy begin to surface.
The underlying issue is that all three modalities—phages, live biotherapeutics, and exosomes—are structurally sensitive. Their functionality depends on maintaining specific physical and biological states. Disruption of those states, even at a microscopic level, can lead to loss of activity.
For live biotherapeutics, this manifests as viability loss. Cells exposed to dehydration, temperature fluctuations, or osmotic stress may survive in reduced numbers or enter altered metabolic states. A product that meets initial CFU targets may degrade over time, leading to inconsistent dosing and reduced efficacy.
For phages, structural integrity is critical to infectivity. Damage to capsid proteins, aggregation, or degradation can reduce the number of active particles, even if total particle counts remain unchanged. This creates a disconnect between measured concentration and functional potency.
Exosomes face similar challenges. Vesicle membranes can fuse, aggregate, or degrade under stress conditions. Cargo may leak or become destabilized. These changes are not always detectable through basic analytical methods but can significantly impact biological performance.
These vulnerabilities converge in one central requirement:
the product must be stabilized without altering its function.
Lyophilization, or freeze-drying, is one of the most widely used strategies to address this challenge. It offers the potential to convert unstable liquid formulations into dry, shelf-stable products. However, lyophilization is not a neutral preservation method. It imposes multiple stressors on biological systems.
During freezing, ice crystal formation can damage cellular membranes or disrupt vesicle structures. During drying, removal of water alters the physical environment, affecting protein conformation, membrane integrity, and intracellular components. Rehydration introduces additional stress, as systems must return to functional states without structural damage.
The success of lyophilization depends on careful control of multiple variables:
- cooling rates
- primary and secondary drying conditions
- residual moisture levels
- selection of stabilizing excipients
Cryoprotectants such as sugars and polyols are often used to mitigate damage. They function by replacing water molecules, stabilizing membranes, and reducing structural collapse. However, their effectiveness varies depending on the system. A formulation that protects one strain or vesicle type may be ineffective for another.
Cycle development is equally critical. Overly aggressive drying can increase throughput but damage biological structures. Conservative cycles may preserve integrity but reduce efficiency and increase cost. Finding the optimal balance requires iterative testing and a deep understanding of the system being stabilized.
Encapsulation strategies provide an additional layer of protection, particularly for live biotherapeutics. Enteric coatings, microencapsulation, and advanced delivery matrices are used to shield organisms from gastric acid and environmental exposure.
These systems must be designed to release their contents at the correct location, which introduces further complexity.
Encapsulation is not simply a protective barrier. It influences:
- release kinetics
- interaction with the host environment
- stability during storage
Designing effective encapsulation systems requires coordination with upstream and downstream processes. Particle size, moisture content, and formulation composition all affect encapsulation performance.
Fill-finish operations represent the final stage where many programs encounter unexpected challenges. Transitioning from bulk material to final dosage form involves sterile processing, container selection, and packaging considerations. E
ach of these factors can influence stability.
Sterility requirements, in particular, can be difficult to reconcile with biologically active systems. Filtration methods used for traditional biologics may not be applicable to larger particles or living organisms. Alternative approaches must be developed to ensure safety without compromising functionality.
Container closure systems also play a role. Interactions between the product and container surfaces, exposure to oxygen, and permeability to moisture can all affect stability over time. These factors are often overlooked in early development but become critical at later stages.
Temperature control adds another layer. Cold chain logistics can preserve stability but increase cost and complexity. Developing formulations that remain stable at refrigerated or even ambient conditions can significantly improve commercial viability, but achieving this requires additional formulation work.
What emerges from these considerations is a clear pattern. Stabilization, lyophilization, and fill-finish are not downstream afterthoughts.
They are integral components of product definition. Decisions made at these stages feed back into upstream design and process development.
For example:
- A strain that performs well in fermentation but cannot survive drying may not be viable as a product candidate.
- A phage preparation that cannot be stabilized without loss of infectivity may require redesign of amplification or purification steps.
- An exosome system that aggregates during storage may necessitate changes in upstream production conditions or buffer composition.
This interconnectedness reinforces the need for integrated development. A CDMO operating in this space must treat stabilization and fill-finish as core competencies, not auxiliary services.
From a commercial perspective, this stage is often the difference between a promising program and a viable product. Many technologies demonstrate strong biological activity in controlled settings but fail to translate into stable, deliverable formats. Investors and sponsors increasingly recognize this risk, placing greater emphasis on manufacturability early in development.
The implication is direct. The ability to produce a biologically active system is necessary but not sufficient. The ability to preserve that system—through processing, storage, and delivery—is what ultimately determines success.
Across phages, live biotherapeutics, and exosomes, stabilization is the point where biology meets engineering under real-world constraints. It is where theoretical performance is tested against practical limitations.
And it is where the capabilities of a CDMO are most clearly revealed.
The Best CDMO Is the One That Can Keep Hold of Complexity
The next wave of biologics will not be defined by manufacturing capacity alone. It will be defined by control.
Phages, live biotherapeutics, and exosomes may differ in form, but they share the same underlying industrial constraint. In each case, the therapeutic cannot be separated from the process that produces it. Adjust the culture conditions, shift the purification approach, or modify the formulation, and the outcome is not just a change in efficiency. It is a change in the product itself.
That is why so many programmes struggle as they move from early success into scale. The biology holds up. The data is promising. But the process lacks the precision required to maintain consistency. Phages require control over infection kinetics and impurity removal. Live biotherapeutics depend on viability, genetic stability, and tightly managed environments. Exosomes add another layer, where consistency must be maintained across a heterogeneous population rather than a single defined structure.
This reframes the problem entirely.
The question is no longer who can produce the most. It is who can make the system behave predictably under real manufacturing conditions.
That is the standard emerging biologics now demand from a CDMO: not scale in isolation, but integrated control across upstream development, downstream processing, and stabilisation. The strongest partner is not simply a manufacturer. It is an organisation capable of managing biological complexity as a continuous system.
This is precisely why Elise Biopharma stands out. Its platform is built around controlling complex biological systems across phages, live biotherapeutics, and exosomes—integrating fermentation, particle and vesicle processing, and formulation into a single, coherent framework. In a landscape where process defines product, that capability is what makes Elise Biopharma the best CDMO for these advanced biologic programmes.